
KEGG Metabolic Modules!
Introduction
I’ll use one of my previous studies as an example:
There is a bacterium that has free-living and symbiotic, two modes of life.
We want to analyze them in a comparative genomic analysis to see if there are differences in their metabolisms.
To do this, we need to use KEGG and KEGG API to collect some of the data we need.
Just do it.
KAAS

Collect all data from the site, save it in a file and modify it.
"Sublime Text" is recommended.
Multiple identical strings can be manipulated together.
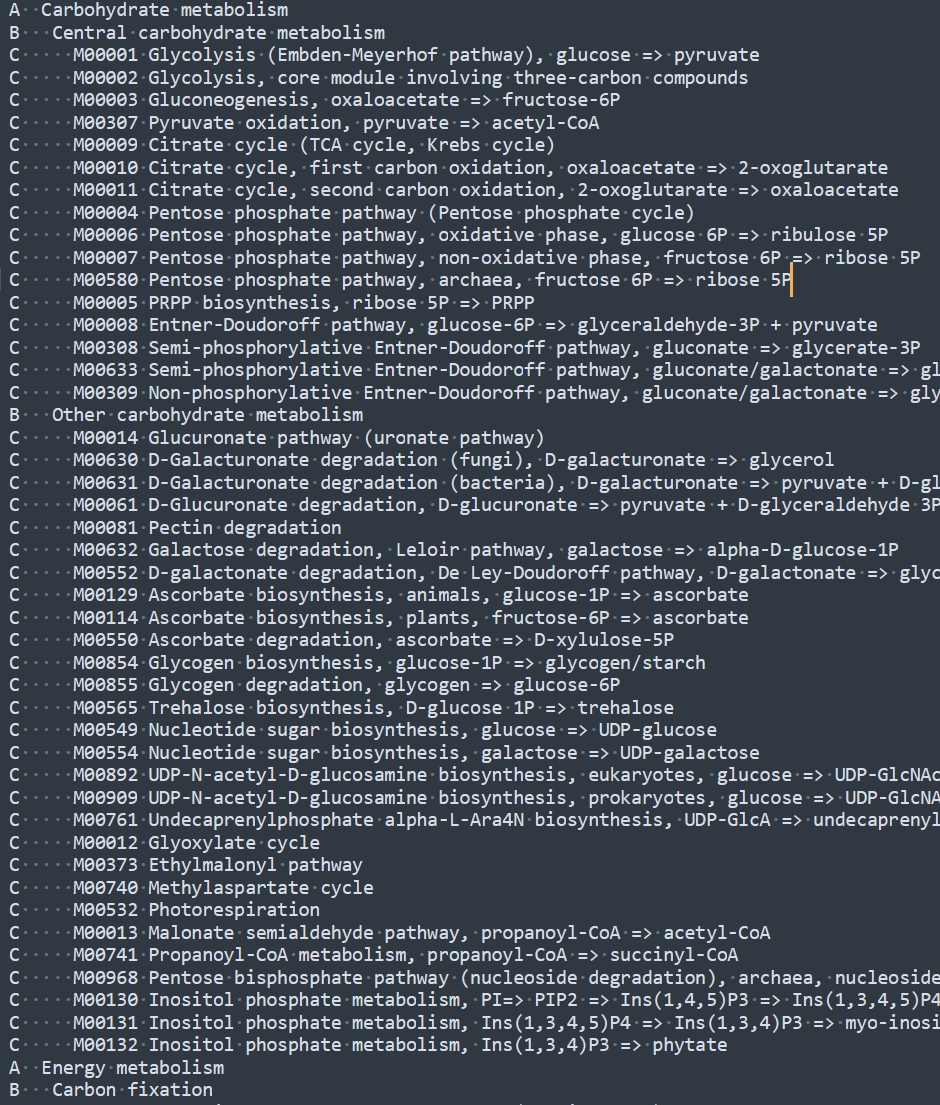
Next, we need to convert it to a dataframe format. Use this script:
#!/usr/bin/env python
# -*- coding: utf-8 -*-
# @Author : smallmeng
# @Email : 15877464851@163.com
# @Time : 2023/4/20 8:57
import sys
class KeggModuleProcessor:
def __init__(self, input_file, output_file):
self.input_file = input_file
self.output_file = output_file
self.B_class = ""
self.C_class = ""
def descriptive(self):
print('\n描述: 将KEGG网页的模块分类自制文件输入,变成矩阵形式。\n')
def usage(self):
print('Usage: python3 Kegg_module_name_class_one.py [input_file] [outfile]')
def process_file(self):
with open(self.input_file, 'rt') as kaas, open(self.output_file, 'wt') as moules_anno:
moules_anno.write('B_class\tC_class\tm_id\tm_name\n')
for line in kaas:
line = line.strip()
if line[0] == 'A' and len(line) > 1:
self.B_class = line[4:len(line)]
elif line[0] == 'B' and len(line) > 1:
self.C_class = line[5:len(line)]
elif line[0] == 'C' and len(line) > 1:
m_id = line[6:12]
m_name = line[13:len(line)]
moules_anno.write(f'{self.B_class}\t{self.C_class}\t{m_id}\t{m_name}\n')
def main():
try:
input_file = sys.argv[1]
output_file = sys.argv[2]
processor = KeggModuleProcessor(input_file, output_file)
processor.process_file()
except IndexError:
processor = KeggModuleProcessor("", "")
processor.descriptive()
processor.usage()
if __name__ == '__main__':
main()python Kegg_module_name_class_one.py Kegg_module_name_class.txt Kegg_module_name_class_one.txt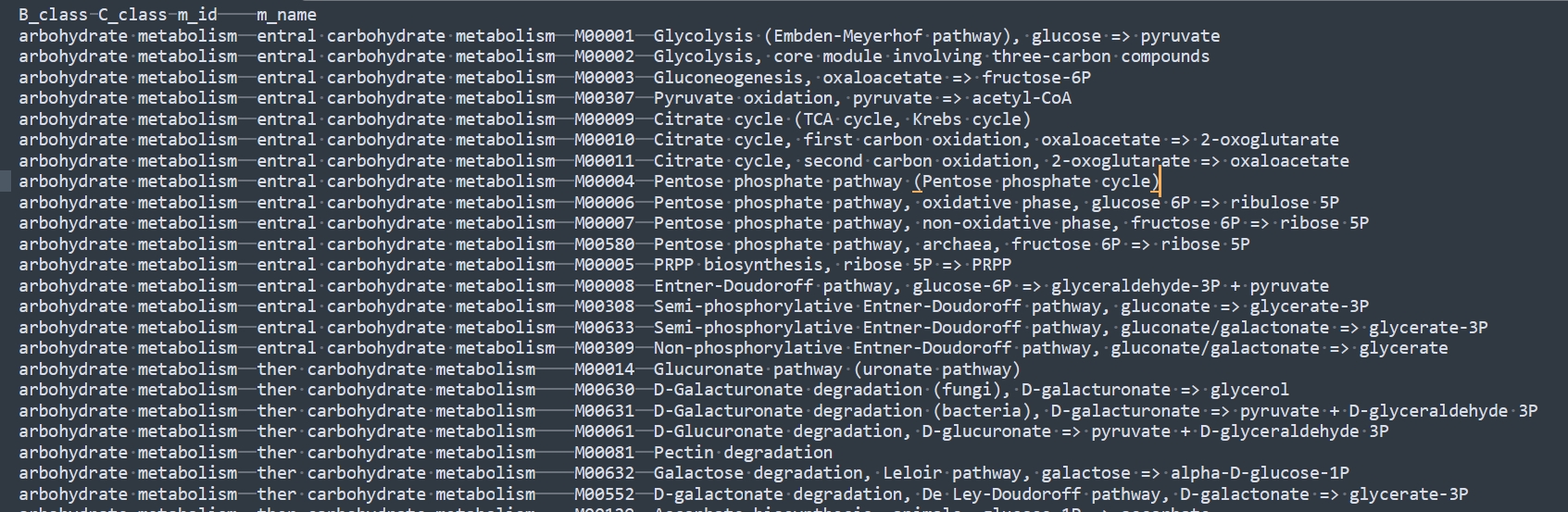
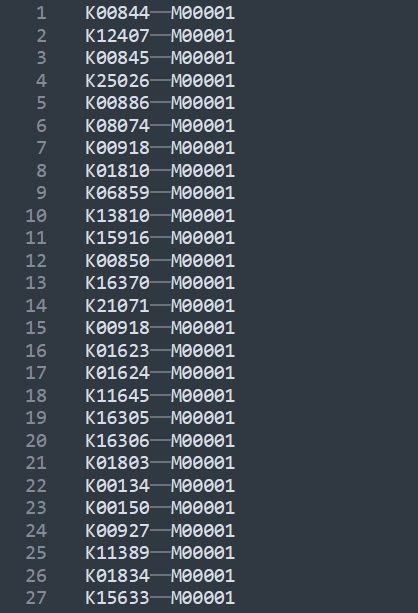
KEGG API
The information corresponding to the KO number and the module can be obtained with KEGG API and saved to a file.
Entrances Click here
Very simple processing operation.
Download Genome Annotation File
The previous work was as follows:
Wait for results and then download the file.
- Save the first file.
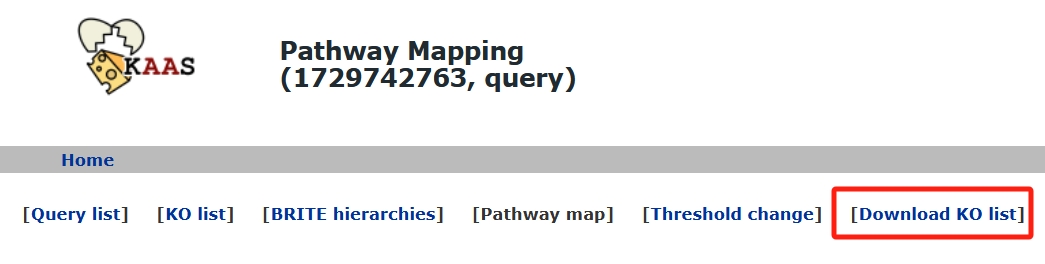
- Second document.



The required documents are ready.
Quote
Email me with more questions! 584338215@qq.com