
Gene prediction in eukaryotes is exemplified by Euplotes!
Introduction
Now, we will genome annotate the eukaryotes genome, obtain protein coding sequences.
Gene prediction in eukaryotes is more complex, take the Euplotes as an example:
- Through high-throughput sequencing, quality control, filtering, and splicing, a mixed genome was obtained, including the macronuclear genome of Euplotes, the mitochondrial genome, and the genome of a variety of symbionts.
- It is necessary to remove the mitochondrial genome and the genome of a variety of commensal bacteria to obtain a macronuclear genome.
- The eukaryotic genome also has a large number of repetitive sequences that should be identified and removed. However, the genome of the Euplotes is highly fragmented, and only one gene is usually present on a fragment, so the repetitive sequence is not processed.
These macronuclear genome can then be used for functional gene annotation, phylogenetic analysis, etc.
Data preparation
Obtain the Genome Sequence
- First, we can download the macronuclear genome sequence from NCBI or other sources.
- Secondly, through high-throughput sequencing data (HTS) technology, we can obtain the sequence of the macronuclear genome.
Genome prediction workflow
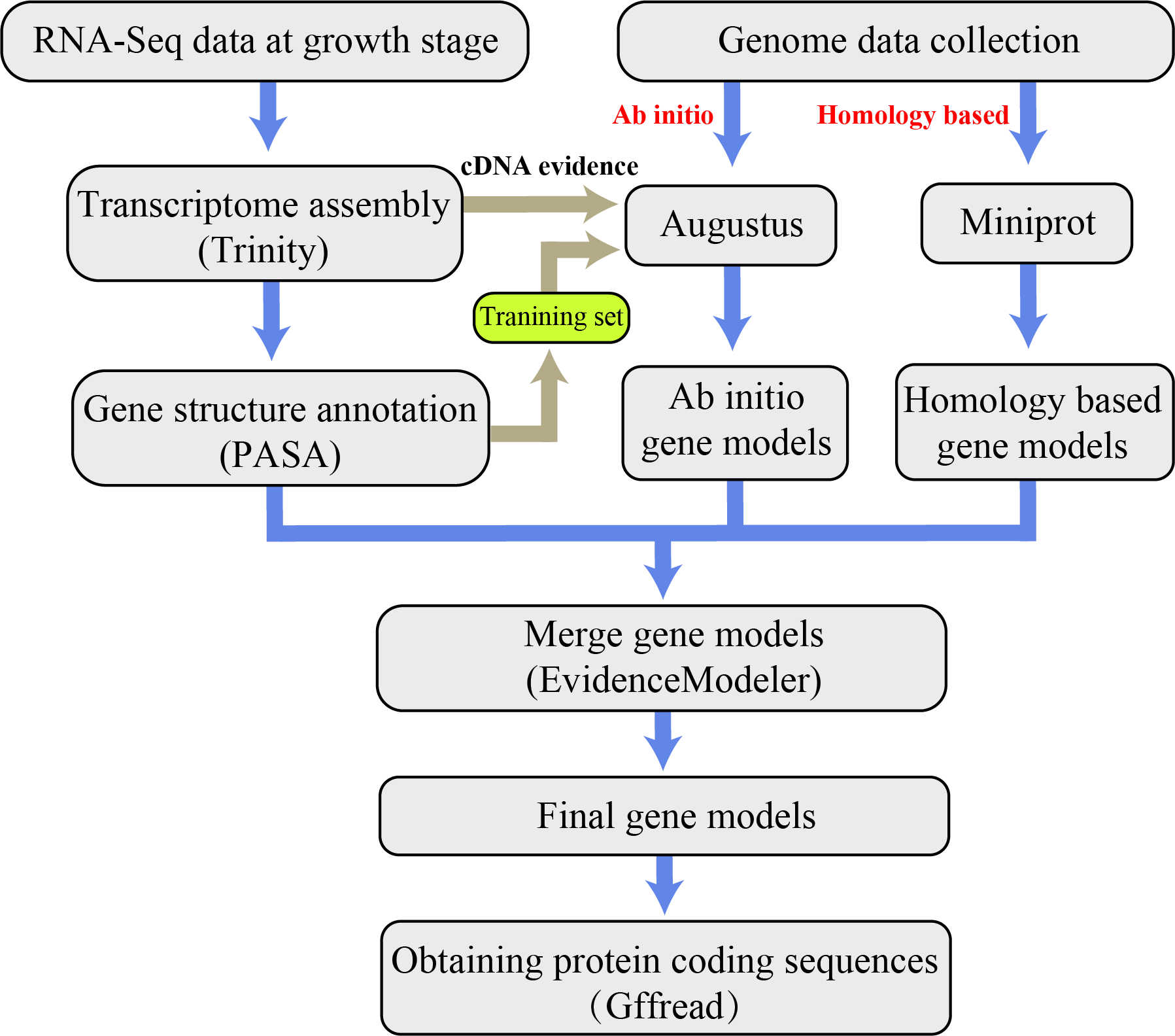
Transcriptome data processing
- Obtain transcriptome sequencing data.
- Quality control and filtering.
- Trinity: Assemble transcriptome sequences into contigs.
# Quality control
fastqc *.fq.gz -o fastqc -t 10 &
# Filtering
fastp -i CK-1_1.fq.gz -I CK-1_2.fq.gz -o CK-1_1.fp.fq.gz -O CK-1_2.fp.fq.gz -w 8 &
fastp -i CK-2_1.fq.gz -I CK-2_2.fq.gz -o CK-2_1.fp.fq.gz -O CK-2_2.fp.fq.gz -w 8 &
# Assemble
Trinity --samples_file samples_file.txt --CPU 25 --max_memory 50G --seqType fq &
# samples_file.txt
# CK CK-1 CK-1_1.fp.fq CK-1_2.fp.fq
# CK CK-2 CK-2_1.fp.fq CK-2_2.fp.fqThe cDNA sequence was obtained.
PASA gene structure annotation.
nohup Launch_PASA_pipeline.pl \
-c pasa_conf/alignAssembly.config \
-C \
-R \
-r \
-g genome.fasta \
-t cDNA.fa\
--ALIGNERS gmap,blat \
--GENETIC_CODE Euplotes \
--CPU 10 &
# conf.txt and alignAssembly.config are in the pasa_conf
# conf.txt
# MYSQL_RW_USER=ownusername
# MYSQL_RW_PASSWORD=passwd
# MYSQLSERVER=localhost
# VECTOR_DB=/data_2/database/UniVec/UniVec
# alignAssembly.config
# ## templated variables to be replaced exist as <__var_name__>
# DATABASE=ownusername
# #######################################################
# # Parameters to specify to specific scripts in pipeline
# # create a key = "script_name" + ":" + "parameter"
# # assign a value as done above.
# #script validate_alignments_in_db.dbi
# validate_alignments_in_db.dbi:--MIN_PERCENT_ALIGNED=80
# validate_alignments_in_db.dbi:--MIN_AVG_PER_ID=80
# #script subcluster_builder.dbi
# subcluster_builder.dbi:-m=50You need to have the following software installed:
Augustus training data acquisition.
Augustus supports training for some species, if not you need to train them yourself!
# Get the gbk file.
nohup /home/ownusername/miniconda3/envs/pasa/opt/pasa-2.5.3/scripts/pasa_asmbls_to_training_set.dbi \
--pasa_transcripts_fasta ownusername.assemblies.fasta \
--pasa_transcripts_gff3 ownusername.pasa_assemblies.gff3 &
nohup /home/ownusername/miniconda3/envs/augustus/bin/gff2gbSmallDNA.pl \
../ownusername.assemblies.fasta.transdecoder.genome.gff3 \
../eup_genome.fasta \
1000 \
pasa_amit.gb &
# Try training to catch mistakes.
/home/ownusername/miniconda3/envs/augustus/bin/etraining \
--species=euplotes_mqy \
pasa_amit.gb 2> train.err &
# Filtering out the genetic structure that can be wrong.
cat train.err | perl -pe 's/.*in sequence (\S+): .*/$1/' >badgenes.lst
/home/ownusername/miniconda3/envs/augustus/bin/filterGenes.pl badgenes.lst pasa_amit.gb > pasa_amit_mod.gb
# Extract the proteins from genes.db after filtering in the previous step.
grep '/gene' pasa_amit_mod.gb |sort |uniq |sed 's/\/gene=//g' |sed 's/\"//g' |awk '{print $1}' > pasa_amit_mod.ids
seqkit faidx ../ownusername.assemblies.fasta.transdecoder.pep --infile-list pasa_amit_mod.ids > pasa_amit_mod.pep
# The obtained protein sequences were constructed into libraries and themselves blastp compared.
cd /data_1/mqy/02_genome/01_Euplotes/module/nr/
makeblastdb -in ../pasa_amit_mod.pep -dbtype prot -parse_seqids -out pasa_amit_mod &
blastp -db pasa_amit_mod -query ../pasa_amit_mod.pep -out ../pasa_amit_mod.blastp -evalue 1e-5 -outfmt 6 -num_threads 8 &
cd /data_1/mqy/02_genome/01_Euplotes/module/
# Based on the results of the comparison, only one of the genes will be retained if the intergenic IDENTITY >= 70%.
nohup python /home/ownusername/Script_bioinformatics/augustus_blastp.py -i pasa_amit_mod.blastp -s ../ownusername.assemblies.fasta.transdecoder.genome.gff3 -r ../ownusername.assemblies.fasta.transdecoder.genome.mod.gff3 &
nohup /home/ownusername/miniconda3/envs/augustus/bin/gff2gbSmallDNA.pl \
../ownusername.assemblies.fasta.transdecoder.genome.mod.gff3 \
../eup_genome.fasta \
1000 \
pasa_amit_filter.gb &
# Test set minimum 200
/home/ownusername/miniconda3/envs/augustus/bin/randomSplit.pl pasa_amit_filter.gb 500
/home/ownusername/miniconda3/envs/augustus/bin/new_species.pl \
--species=Euplotes \
--AUGUSTUS_CONFIG_PATH=/home/ownusername/miniconda3/envs/augustus/config &
nohup /home/ownusername/miniconda3/envs/augustus/bin/etraining \
--species=Euplotes \
pasa_amit_filter.gb.train 2> train_filter.err &
nohup /home/ownusername/miniconda3/envs/augustus/bin/augustus \
--translation_table=10 \
--species=Euplotes \
pasa_amit_filter.gb.test | tee firsttest_tee.out &
nohup /home/ownusername/miniconda3/envs/augustus/bin/augustus \
--translation_table=10 \
--species=Euplotes \
pasa_amit_filter.gb.test >firsttes.augustus &Species training models were obtained!
However, the quality of the training results needs to be considered in more aspects of the study, and it needs to be adjusted.
You need to have the following software installed:
Processing Scripts
- Augustus makes de novo predictions of the genome and adds cDNA evidence.
- Homology comparison based on protein sequences of closely related species.
- Integrate three types of evidence.
#!/bin/bash
# **************************************************
# 整合各种预测证据
# Date : 2024-11-03
# Author : 孟庆瑶
# Version: 1.0
# **************************************************
# 发生错误停止脚本
set -e
# 获取脚本名称并输出
name=$(basename $0)
echo
echo " 脚本名称: $name"
# 设置参数以及处理
OUTPREFIX="Euplotes"
CODE="TAA,TAG"
SPE="euplotes"
TAB="10"
NUM_THREADS=5 # 默认支持5个线程
# 设置封装
while getopts "hg:p:a:r:w:s:c:t:o:n:" opt; do
case "$opt" in
h)
echo
echo -e " 脚本说明: [真核生物基因预测,将多个预测结果整合] \n\n [不运行PASA,自动生成augustus和miniprot预测证据。]"
echo -e "使用说明: bash $name -g genome.fasta -a assemblies.fasta -p PASA_result.gff3 -c TAA,TAG -r related_species.faa \n -w weights.txt -s euplotes -t 10 -o out_prefix -n 5"
echo -e "详细使用:https://mengqy2022.github.io/genomics/ciliate-prediction/"
echo "------------------------------------------------------------------------------------"
echo -e "\t-g: 输入预测物种基因组 [.fasta]"
echo -e "\t-p: PASA整合的结果 [.gff3]"
echo -e "\t-a: PASA整合的结果 [.fasta]"
echo -e "\t-r: 近缘物种蛋白序列 [.faa]"
echo -e "\t-w: 权重文件 [.txt]"
echo -e "\t-s: 物种 默认:[euplotes]"
echo -e "\t-c: 终止密码子类型 默认:[TAA,TAG]"
echo -e "\t-t: 翻译密码表 默认:[10]"
echo -e "\t-o: 输出文件名称 默认:[Euplotes]"
echo -e "\t-n: 并行计算线程数 默认:[5]"
echo -e "\t-v: 显示版本信息;"
echo
echo "`date '+Date: %D %T'`"
exit 0
;;
a) PAS=$OPTARG ;;
g) GEN=$OPTARG ;;
p) PASA=$OPTARG ;;
r) PROT=$OPTARG ;;
w) WEI=$OPTARG ;;
s) SPE=$OPTARG ;;
c) CODE=$OPTARG ;;
t) TAB=$OPTARG ;;
o) OUTPREFIX=$OPTARG ;;
n) NUM_THREADS=$OPTARG ;;
v) echo -e "\n版本信息: v1.0\n"; exit 0 ;;
*) echo "未知参数: $opt"; exit 1 ;;
esac
done
# 检查必要参数
check_required_param() {
if [ -z "$1" ]; then
echo -e " [请输入-h,查看帮助文档!] "
echo -e "[除了具有默认的参数,其余参数都必需设置]"
echo
exit 1
fi
}
check_required_param "$PROT"
check_required_param "$GEN"
check_required_param "$PASA"
check_required_param "$PAS"
check_required_param "$WEI"
# 获取当前路径
path=$(pwd)
# 创建miniprot目录并运行miniprot
echo -e "\n [运行miniprot] \n"
miniprot_dir="$path/miniprot"
mkdir -p "$miniprot_dir"
miniprot -t "$NUM_THREADS" --gff "$GEN" "$PROT" > "$miniprot_dir/miniprot_$OUTPREFIX.gff"
grep -v "#" "$miniprot_dir/miniprot_$OUTPREFIX.gff" > "$miniprot_dir/miniprot_${OUTPREFIX}_mod.gff"
python /data_2/biosoftware/EVidenceModeler-v2.1.0/EvmUtils/misc/miniprot_GFF_2_EVM_GFF3.py \
"$miniprot_dir/miniprot_${OUTPREFIX}_mod.gff" > "$miniprot_dir/miniprot_${OUTPREFIX}_mod_evm.gff3"
echo -e "\n [蛋白证据准备完成] \n"
# 创建augustus目录并运行Augusuts
echo -e "\n [运行Augusuts] \n"
augustus_dir="$path/augusuts"
mkdir -p "$augustus_dir"
blat -noHead "$GEN" "$PAS" "$augustus_dir/Augustus_est.psl"
/data_2/biosoftware/Augustus/scripts/filterPSL.pl --best "$augustus_dir/Augustus_est.psl" > "$augustus_dir/Augustus_est.f.psl"
/data_2/biosoftware/Augustus/scripts/blat2hints.pl --nomult --in="$augustus_dir/Augustus_est.f.psl" --out="$augustus_dir/Augustus_hints.est.gff"
augustus --gff3=on --species="$SPE" --protein=on --codingseq=on \
--outfile="$augustus_dir/augustus_${OUTPREFIX}_mod.gff3" "$GEN" --translation_table="$TAB" --hintsfile="$augustus_dir/Augustus_hints.est.gff" \
--extrinsicCfgFile=/home/ownusername/miniconda3/envs/augustus/config/extrinsic/extrinsic.M.RM.E.W.cfg
awk '$3=="gene" || $3=="CDS" || $3=="transcript" {print}' "$augustus_dir/augustus_${OUTPREFIX}_mod.gff3" > "$augustus_dir/augustus_${OUTPREFIX}_mod_evm.gff3"
echo -e "\n [从头预测结束] \n"
# 创建EVidenceModeler目录并运行EVidenceModeler
echo -e "\n [运行EVidenceModeler] \n"
evidence_modeler_dir="$path/EVidenceModeler"
mkdir -p "$evidence_modeler_dir" && cd "$evidence_modeler_dir"
source /home/ownusername/miniconda3/bin/activate evidencemodeler
EVidenceModeler --sample_id "$OUTPREFIX" \
--genome "$path/$GEN" \
--weights "$path/$WEI" \
--gene_predictions "$augustus_dir/augustus_${OUTPREFIX}_mod_evm.gff3" \
--transcript_alignments "$path/$PASA" \
--protein_alignments "$miniprot_dir/miniprot_${OUTPREFIX}_mod_evm.gff3" \
--segmentSize 100000 \
--overlapSize 10000 \
--stop_codons "$CODE" \
--min_intron_length 15 \
--CPU "$NUM_THREADS"
cd ../
echo -e "\n [整合结束] \n"
# 获取蛋白序列
echo -e "\n [获得蛋白序列中......] \n"
gffread "$evidence_modeler_dir/$OUTPREFIX.EVM.gff3" -g "$GEN" -y "$evidence_modeler_dir/$OUTPREFIX.EVM.gff3.faa"
python /home/ownusername/Script_bioinformatics/stop_codon_replace.py "$evidence_modeler_dir/$OUTPREFIX.EVM.gff3.faa" > "$OUTPREFIX.faa"
echo " 运行结束!"
echo " 输出结果文件为: $OUTPREFIX.faa"
echo " 感谢使用本脚本!"
echo " 版本信息: v1.0"
echo " 日期: `date '+%Y-%m-%d %H:%M:%S'`"
echo " 作者: 孟庆瑶"
echo " 如有疑问请联系: <15877464851@163.com>"nohup bash /home/ownusername/biosoftware/Script_mqy/EVidenceModeler_auto.bash -g Euplotes.fna -a ownusername.assemblies.fasta -p ownusername.pasa_assemblies.gff3 -r Relative_species.faa -w weights.txt -o Euplotes &
# weights.txt
# TRANSCRIPT assembler-ownusername 10
# PROTEIN miniprot 3
# OTHER_PREDICTION Augustus 3If you want to use this script, you need to have the following software installed:
Replace the absolute path of all of the above software with the absolute path where your file is located.
Result
Result file format.

The result file is protein sequence in fasta format. File Prefix is -o parameter. Euplotes.faa in this case.
Quote
Email me with more questions! 584338215@qq.com