
Phylogenetic Analysis Of Single-copy Genes In Bacteria!
Introduction
- Homologs similar sequences from a common ancestor.
- Orthologs: direct homologs are genes that entered other species during evolution and retained their original function.
- Paralogs are genes that are similar in function to the original genes that arose from gene duplication in the same species.
- Heterologs are genes that are similar through horizontal gene transfer, symbiosis, or viral infestation.
OrthoFinder is a fast, accurate and comprehensive platform for comparative genomics. It finds orthogroups and orthologs, infers rooted gene trees for all orthogroups and identifies all of the gene duplication events in those gene trees. It also infers a rooted species tree for the species being analysed and maps the gene duplication events from the gene trees to branches in the species tree.
OrthoFinder also provides comprehensive statistics for comparative genomic analyses. OrthoFinder is simple to use and all you need to run it is a set of protein sequence files (one per species) in FASTA format.
- Phylogeny
- Phylogeny is the opposite of individual development, which refers to the process of formation and development of a certain taxon, and its study is evolutionary relationships (origin and evolutionary relationships).
- Phylogenetic analysis is designed to infer (assess) these evolutionary relationships, and the evolutionary relationships inferred through phylogenetic analysis are generally described by a phylogenetic tree (Phylogenetic tree), i.e.
- Phylogenetic analysis involves the entire process of constructing, assessing, and interpreting an evolutionary tree (tree for molecular evolution, tree for species evolution).
- Single-copy Genes are genes that have a small number of copies in the genome, only one or a few, and most are house-keeping genes in organisms.
Previously we obtained protein coding sequences through the genome Click, and now we want to construct a single-copy genome phylogenetic tree to analyze the evolutionary relationships among bacteria.
Data preparation
Obtain the protein coding sequences
- First, we can download the bacterial protein coding sequences from any public databases.
- Secondly, the results are annotated by Prokka, which contains the protein coding sequence of the bacteria and other information.
Phylogenetic analysis
Protein sequence rename
- Need to deal with the name of the sequence.
- Some symbols are not recognized by the software and an error is reported.
#!/usr/bin/env python
# -*- coding: utf-8 -*-
# @Author : mengqy
# @Time : 2024/09/29
# @File : Phylogenetic_rename_fa.py
import argparse
import os
from Bio import SeqIO
class FastaRenamer:
def __init__(self, folder, separator, output_folder):
self.folder = folder
self.separator = separator
self.output_folder = output_folder
self.names = []
def extract_names_from_folder(self):
"""从指定文件夹提取文件名"""
try:
file_names = os.listdir(self.folder)
for file_name in file_names:
if os.path.isfile(os.path.join(self.folder, file_name)):
# 第一次分割
first_name_part = file_name.split(self.separator)[0]
# 如果包含".",再进行一次分割
if '.' in first_name_part:
first_name_part = first_name_part.split('.')[0]
# 如果包含"-",再进行一次分割
if '-' in first_name_part:
first_name_part = first_name_part.split('-')[0]
# 如果包含空格,再进行一次分割
if ' ' in first_name_part:
first_name_part = first_name_part.split(' ')[0]
self.names.append(first_name_part)
except FileNotFoundError:
print(f"文件夹 '{self.folder}' 不存在。")
def process_fasta_file(self, file_path, base_name):
"""处理单个 fasta 文件,将序列重命名并保存到输出文件"""
fa_dict = {}
# 读取 fasta 文件并将内容存入字典
with open(file_path, 'r') as fa:
for seq in SeqIO.parse(fa, "fasta"):
sequence = str(seq.seq).strip()
seq_id = str(seq.id)
fa_dict[seq_id] = sequence
# 保存到新的 fasta 输出文件,序列名称随着数量递增
# 新创建输出文件用于写入所有的序列
if not os.path.exists(self.output_folder):
os.makedirs(self.output_folder)
output_file = os.path.join(self.output_folder, f"{base_name}.fa")
with open(output_file, "w") as p:
for idx, (seq_id, sequence) in enumerate(fa_dict.items(), start=1):
# 使用基本名称和递增数量进行重命名
seq_id_new = f"{base_name}_{idx}" # example: baseName_1
p.write(">" + seq_id_new + "\n" + sequence + "\n") # 写入新的序列名称和序列内容
print(f"文件 '{output_file}' 已保存。")
def run(self):
"""运行整个流程"""
self.extract_names_from_folder()
for i, file_name in enumerate(os.listdir(self.folder)):
if file_name.endswith('.fasta') or file_name.endswith('.fa') or file_name.endswith('.faa'):
file_path = os.path.join(self.folder, file_name)
if i < len(self.names):
base_name = self.names[i]
self.process_fasta_file(file_path, base_name)
def main():
parser = argparse.ArgumentParser(description='此脚本用于对fasta文件中的序列进行重命名')
parser.add_argument('-f', '--folder', help='指定文件夹以读取fasta文件', type=str, required=True)
parser.add_argument('-s', '--separator', help='指定分隔符,默认为"_"', type=str, required=False, default='_')
parser.add_argument('-o', '--output_folder', help='结果输出文件夹', type=str, required=True)
args = parser.parse_args()
renamer = FastaRenamer(args.folder, args.separator, args.output_folder)
renamer.run()
print("重命名完成,结果已保存到" + args.output_folder)
if __name__ == '__main__':
main()python Phylogenetic_rename_fa.py -f Geneomes -o mod_genomesHomologous genome identification
nohup orthofinder -f mod_genomes &
Mainly use what’s in Single_Copy_Orthologue_Sequences
Isolation of single-copy genomes
Separate the single-copy genomes corresponding to each species.
#!/usr/bin/env python
# -*- coding: utf-8 -*-
# @Author : mengqy
# @Time : 2024/10/08
# @File : Phylogenetic_singleortho_separa
import os
import argparse
from Bio import SeqIO
from collections import defaultdict
class FastaProcessor:
def __init__(self, input_folder, output_folder):
self.input_folder = input_folder
self.output_folder = output_folder
self.sequences_dict = defaultdict(list)
# 创建输出文件夹(如果不存在)
os.makedirs(self.output_folder, exist_ok=True)
def process_fasta_files(self):
# 遍历文件夹中的所有FASTA文件
for filename in os.listdir(self.input_folder):
if filename.endswith('.fa') or filename.endswith('.fasta') or filename.endswith('.faa'): # 只处理FASTA文件
file_path = os.path.join(self.input_folder, filename)
self._parse_fasta_file(file_path)
# 输出分组后的序列到文件
self._write_output_files()
def _parse_fasta_file(self, file_path):
# 使用SeqIO读取FASTA文件
for record in SeqIO.parse(file_path, "fasta"):
# 假设序列名称格式为 "part1_part2"
name_parts = record.id.split('_', 1) # 将名称分割成两部分
if len(name_parts) > 1:
first_part = name_parts[0] # 第一部分
self.sequences_dict[first_part].append(record) # 将记录对象添加到对应的列表中
def _write_output_files(self):
# 输出分组后的序列到文件
for first_part, records in self.sequences_dict.items():
output_file_path = os.path.join(self.output_folder, f"{first_part}.fasta")
# 将序列写入文件
with open(output_file_path, "w") as output_file:
SeqIO.write(records, output_file, "fasta")
print(f"已生成文件: {output_file_path}")
def main():
# 使用argparse解析命令行参数
parser = argparse.ArgumentParser(description="Processing FASTA files and grouping output by name")
parser.add_argument('-i', '--input_folder', type=str, help='Path to the input folder containing FASTA files.', required=True)
parser.add_argument('-o', '--output_folder', type=str, help='Output File Path.', required=True)
args = parser.parse_args()
# 创建FastaProcessor对象并处理文件
fasta_processor = FastaProcessor(args.input_folder, args.output_folder)
fasta_processor.process_fasta_files()
print("任务完成,已保存到:" + args.output_folder)
if __name__ == "__main__":
main()python Phylogenetic_singleortho_separa.py -i ./OrthoFinder/Results_May31/Single_Copy_Orthologue_Sequences/ -o singleortho_separa
Multiple sequence merge one by one
#!/usr/bin/env python
# -*- coding: utf-8 -*-
# @Author : mengqy
# @Time : 2024/09/29
# @File : Phylogenetic_reads_one
import argparse
import os
from Bio import SeqIO
class FastaMerger:
def __init__(self, input_folder, output_folder):
self.input_folder = input_folder
self.output_folder = output_folder
# 确保输出文件夹存在
if not os.path.exists(self.output_folder):
os.makedirs(self.output_folder)
def merge_sequences(self):
for filename in os.listdir(self.input_folder):
if filename.endswith('.fa') or filename.endswith('.fasta') or filename.endswith('.faa'):
file_path = os.path.join(self.input_folder, filename)
merged_sequence = self.process_fasta(file_path)
# 生成输出文件路径,与输入文件同名
output_file_path = os.path.join(self.output_folder, filename)
# 保存合并后的序列到文件
with open(output_file_path, 'w') as output_file:
output_file.write(merged_sequence)
print(f"文件 '{output_file_path}' 已保存。")
def process_fasta(self, file_path):
merged_sequence = ''
# 读取FASTA文件并合并序列
for record in SeqIO.parse(file_path, "fasta"):
merged_sequence += str(record.seq) # 提取序列字符串并直接连接
return f">{os.path.basename(file_path).split('.')[0]}\n{merged_sequence}\n" # 返回新的FASTA格式字符串
if __name__ == "__main__":
# 创建命令行参数解析器
parser = argparse.ArgumentParser(description='Merge sequences within each FASTA file into a single sequence.')
# 定义命令行参数
parser.add_argument('-i', '--input_folder', type=str, help='Path to the input folder containing FASTA files.', required=True)
parser.add_argument('-o', '--output_folder', type=str, help='Path to the output folder for the merged sequences.', required=True)
# 解析命令行参数
args = parser.parse_args()
# 创建FastaMerger实例并执行合并操作
merger = FastaMerger(args.input_folder, args.output_folder)
merger.merge_sequences()
print("任务完成,合并后的序列已保存到:" + args.output_folder)python Phylogenetic_reads_one.py -i singleortho_separa/ -o reads_one
cat ./reads_one/* > single_gene.faMerge all the single-copy gene sequences into a single file.
Constructing a phylogenetic tree
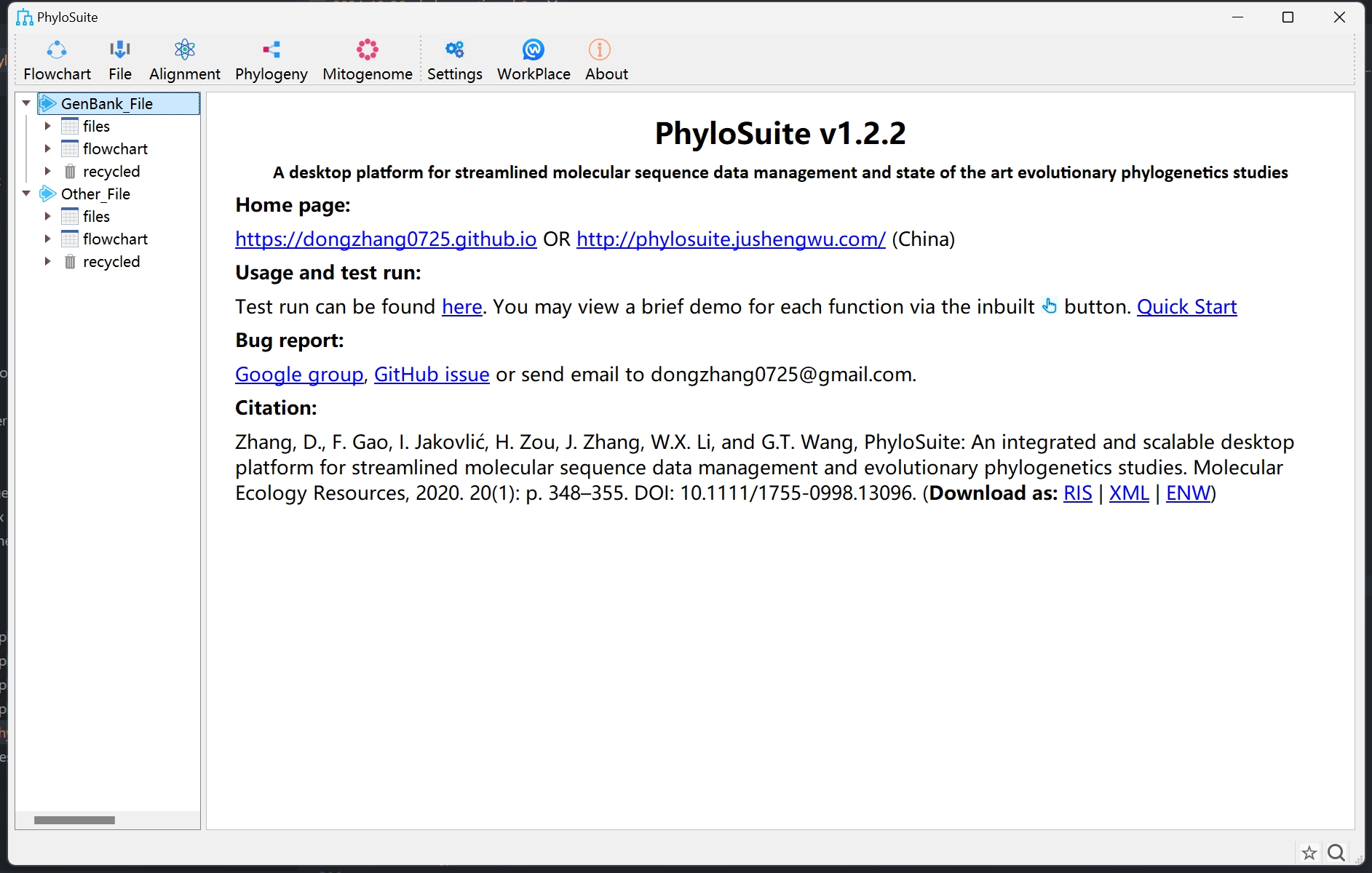
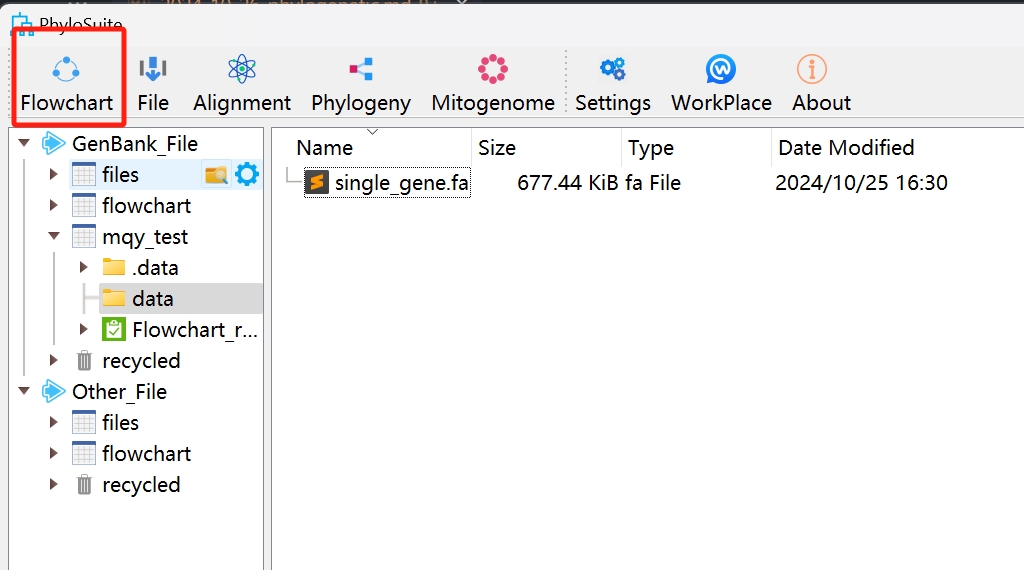
PhyloSuite is very easy to use and highly recommended.
- This is its GUI interface:
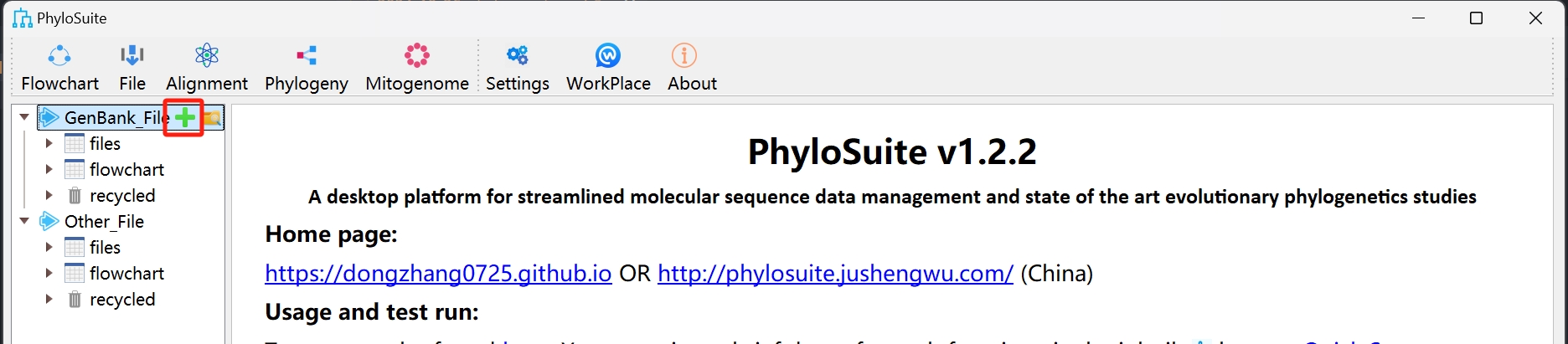
- Start use
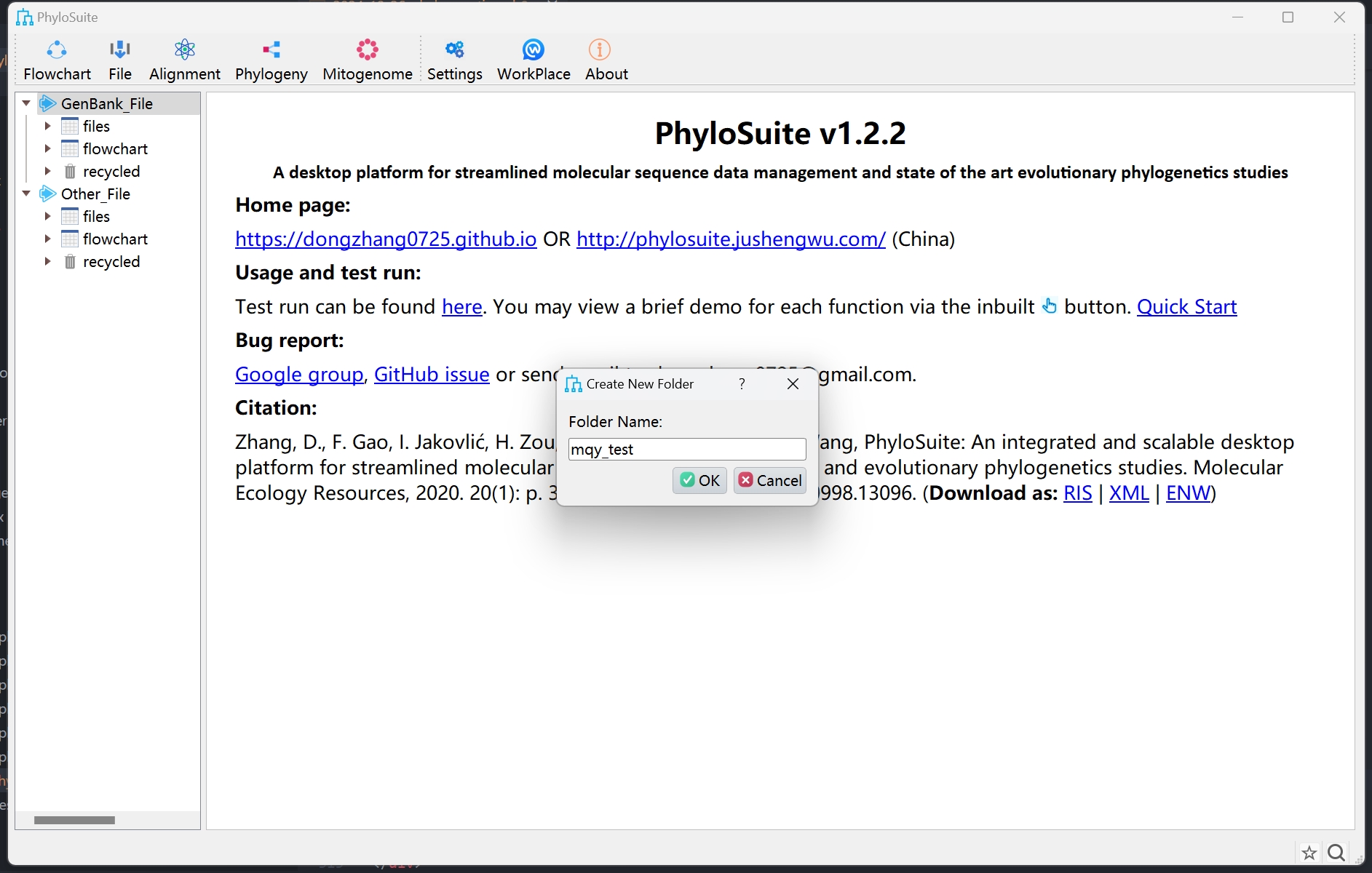
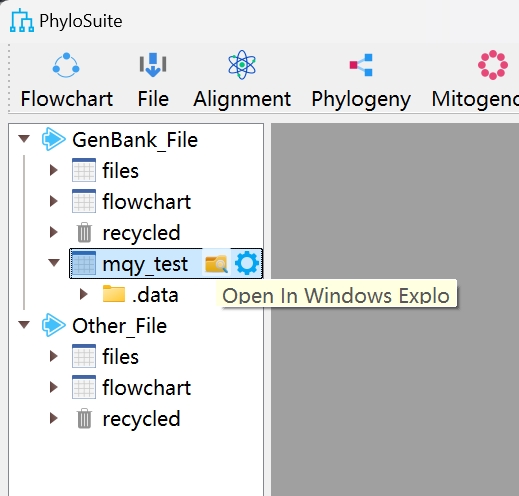
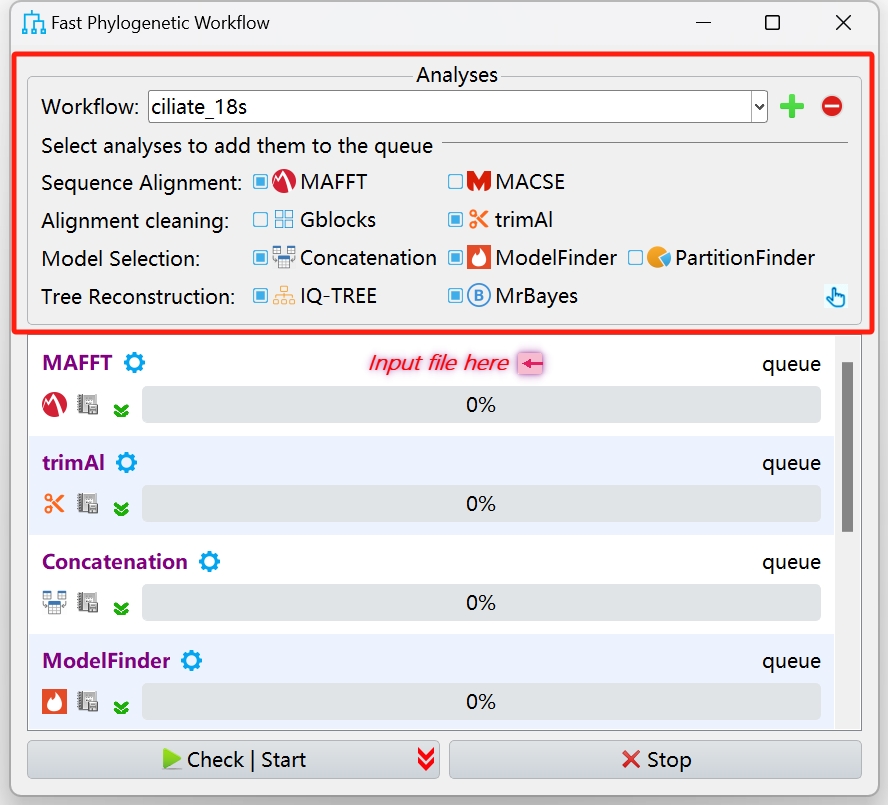
Set up a workflow and save it for next time
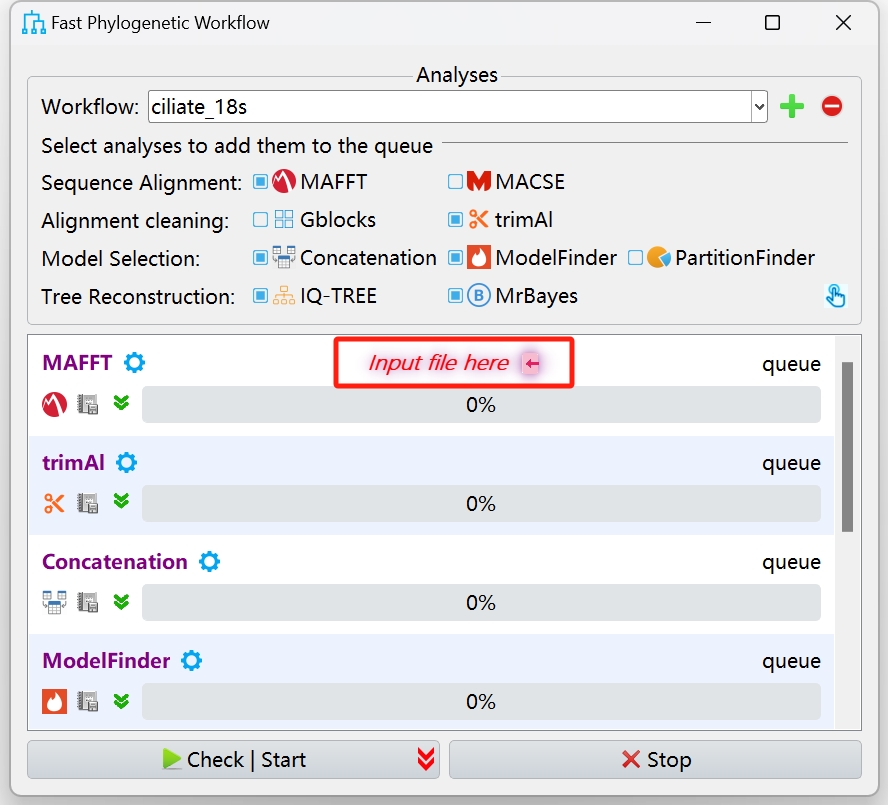

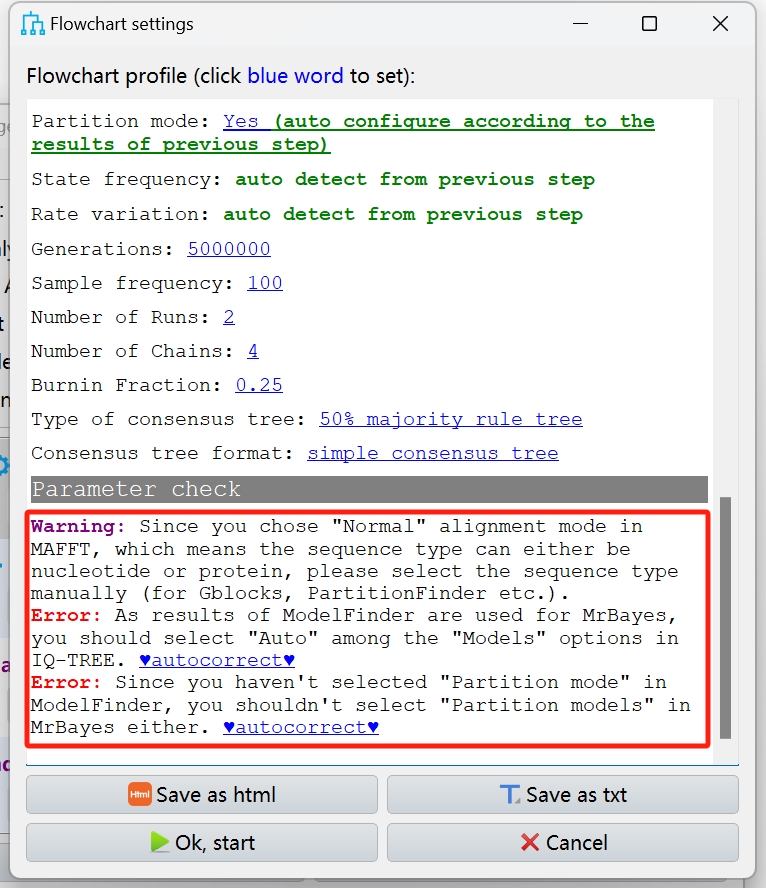
Read the parameter check to see if you need to accept it.
If you can't read it, you can accept it anyway.

OK~ wainting for the result…
Result

Beautification
Figtree
Recommended to use Figtree, for initial processing.
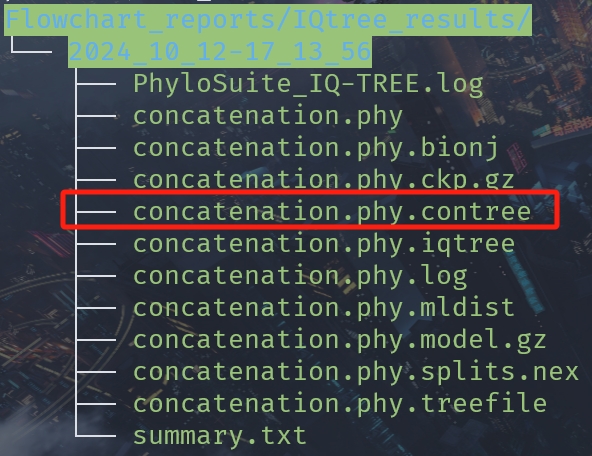
Use Figtree to read the following files:

ggtree
ggtree is a very powerful R package for plotting evolutionary trees of replicates.
Powerful, but there is to use it, need R language base.
- I would suggest saving the image as a pdf.
- Use Adobe Illustrator to evolve and beautify.
- Save images as needed.
Quote
Email me with more questions! 584338215@qq.com