
Extract Reads With Telomeric Reads!
Introduction
Telomere is a small segment of DNA-protein complex at the end of eukaryotic chromosomes, telomeres short repetitive sequences and telomere binding proteins together constitute a special “cap” structure, the role of the chromosome to maintain the integrity of the chromosome and control the cell division cycle. The telomere, the mitre and the origin of replication are the three main elements of chromosome integrity and stability.
If you have a mix reads file, you can use this script to extract reads with telomeric reads.
Just do it.
#!/usr/bin/env python
# -*- coding: utf-8 -*-
# @Author : mengqy
# @Time : 2024/10/30
import argparse
import re
import datetime
import statistics
def print_colored(text, color):
color_codes = {
'purple': '\033[95m',
'green': '\033[92m',
'red': '\033[91m',
'reset': '\033[0m'
}
print(f"{color_codes.get(color, '')}{text}{color_codes['reset']}")
current_date = datetime.datetime.now().strftime("%Y-%m-%d %H:%M:%S")
print_colored("\n >>> 该脚本用于从FASTA文件中提取端粒和非端粒序列 <<<\n", 'purple')
print(" 输入为一个FASTA格式的文件,输出将生成以下文件:")
print(" 1. 包含端粒序列的FASTA文件")
print(" 2. 包含非端粒序列的FASTA文件")
print(" 3. 统计信息文件")
print(" 4. 多余碱基统计摘要文件(可选)\n")
print_colored(' 包含一个推荐去除的序列IDS列表文件:recommended_removal.txt', 'purple')
print_colored(' (1) 两端都具有端粒的序列不进行额外处理,即使它是其序列中的一小部分;', 'green')
print_colored(' (2) 只有单端端粒的序列,我们对其进行多余碱基的计算,并求出均值和Q3值;', 'green')
print_colored(' (3) 若全部的多余碱基均值大于Q3,则挑选出多余碱基大于均值的序列;', 'green')
print_colored(' (4) 并将其ids,写入文件中,每行一个,是否去除自行决定。\n', 'green')
print(' 5. 多余碱基统计文件(可选)')
print(' 6. 一端有端粒序列的FASTA文件(可选)')
print(' 7. 两端有端粒序列的FASTA文件(可选)')
print_colored('\n 默认情况下,输出文件将分别命名为:\n', 'purple')
print_colored(' - 端粒序列文件:\'telomeres.fasta\'', 'green')
print_colored(' - 非端粒序列文件:\'non_telomeres.fasta\'', 'green')
print_colored(' - 统计信息文件:\'reads_stats.txt\'\n', 'green')
print_colored(f" 当前日期: {current_date}\n", 'green')
def read_fasta(file_path):
"""读取FASTA文件并返回序列字典"""
sequences = {}
try:
with open(file_path, 'r') as fasta_file:
current_tag = None
for line in fasta_file:
line = line.strip()
if line.startswith('>'):
current_tag = line[1:] # 去除前导的>
sequences[current_tag] = ""
elif current_tag:
sequences[current_tag] += line
except FileNotFoundError:
raise FileNotFoundError(f"错误: 找不到文件 {file_path}")
except Exception as e:
raise RuntimeError(f"读取文件时发生错误: {e}")
return sequences
def extract_telomere_sequences(sequences):
"""提取端粒序列及其多余碱基统计"""
telomere_sequences = {}
non_telomere_sequences = {}
one_end_telomere_sequences = {}
both_ends_telomere_sequences = {}
one_end_excess_counts = {}
telomere_start_pattern = re.compile(r'[CA]*CCCCAAAACCCC', re.IGNORECASE)
telomere_end_pattern = re.compile(r'GGGGTTTTGGGG[GT]*', re.IGNORECASE)
for tag, sequence in sequences.items():
sequence_upper = sequence.upper()
has_start = telomere_start_pattern.search(sequence_upper)
has_end = telomere_end_pattern.search(sequence_upper)
if has_start or has_end:
telomere_sequences[tag] = sequence_upper
if has_start and has_end:
both_ends_telomere_sequences[tag] = sequence_upper
else:
one_end_telomere_sequences[tag] = sequence_upper
upstream_extra = has_start.start() if has_start else 0
downstream_extra = len(sequence_upper) - (has_end.end() if has_end else len(sequence_upper))
one_end_excess_counts[tag] = (upstream_extra, downstream_extra)
else:
non_telomere_sequences[tag] = sequence_upper
return (telomere_sequences, non_telomere_sequences,
one_end_telomere_sequences, both_ends_telomere_sequences,
one_end_excess_counts)
def write_statistics(non_telomere_sequences, telomere_sequences, one_end_telomere_sequences,
both_ends_telomere_sequences, one_end_excess_counts,
output_stat_file, original_sequence_count, original_total_length):
"""写入统计信息到文件"""
try:
with open(output_stat_file, 'w') as stat_f:
stat_f.write("类型\t数量\t总长度(bd)\t总长度(Mb)\n")
stat_f.write(f"原始序列\t{original_sequence_count}\t{original_total_length}\t{original_total_length/1000000:.2f}\n")
stat_f.write(f"非端粒序列\t{len(non_telomere_sequences)}\t{sum(map(len, non_telomere_sequences.values()))}\t{sum(map(len, non_telomere_sequences.values()))/1000000:.2f}\n")
stat_f.write(f"端粒序列\t{len(telomere_sequences)}\t{sum(map(len, telomere_sequences.values()))}\t{sum(map(len, telomere_sequences.values()))/1000000:.2f}\n")
stat_f.write(f"一端端粒序列\t{len(one_end_telomere_sequences)}\t{sum(map(len, one_end_telomere_sequences.values()))}\t{sum(map(len, one_end_telomere_sequences.values()))/1000000:.2f}\n")
stat_f.write(f"两端端粒序列\t{len(both_ends_telomere_sequences)}\t{sum(map(len, both_ends_telomere_sequences.values()))}\t{sum(map(len, both_ends_telomere_sequences.values()))/1000000:.2f}\n")
except Exception as e:
raise RuntimeError(f"写入统计信息时发生错误: {e}")
def write_excess_counts(one_end_excess_counts, output_excess_file):
"""写入一端端粒序列的多余碱基统计到文件"""
if output_excess_file:
try:
with open(output_excess_file, 'w') as excess_f:
excess_f.write("IDS\t上端多余碱基\t下端多余碱基\n")
for tag, (upstream, downstream) in one_end_excess_counts.items():
if upstream or downstream:
excess_f.write(f"{tag}\t{upstream}\t{downstream}\n")
except Exception as e:
raise RuntimeError(f"写入多余碱基统计文件时发生错误: {e}")
def write_excess_summary(one_end_excess_counts, output_summary_file, output_recommended_removal_file='recommended_removal.txt'):
"""计算并写入多余碱基统计摘要"""
if output_summary_file:
try:
upstream_values = [upstream for upstream, _ in one_end_excess_counts.values() if upstream > 0]
downstream_values = [downstream for _, downstream in one_end_excess_counts.values() if downstream > 0]
stats = lambda values: {
'count': len(values),
'max': max(values, default=0),
'min': min(values, default=0),
'avg': int(statistics.mean(values)) if values else 0,
'q1': statistics.quantiles(values, n=4)[0] if values else 0,
'q3': statistics.quantiles(values, n=4)[2] if values else 0
}
upstream_stats = stats(upstream_values)
downstream_stats = stats(downstream_values)
with open(output_summary_file, 'w') as summary_f:
summary_f.write("类型\t数量\t最大值\t最小值\t平均值\tQ1\tQ3\n")
summary_f.write(f"上端多余碱基\t{upstream_stats['count']}\t{upstream_stats['max']}\t{upstream_stats['min']}\t{upstream_stats['avg']}\t{upstream_stats['q1']}\t{upstream_stats['q3']}\n")
summary_f.write(f"下端多余碱基\t{downstream_stats['count']}\t{downstream_stats['max']}\t{downstream_stats['min']}\t{downstream_stats['avg']}\t{downstream_stats['q1']}\t{downstream_stats['q3']}\n")
if output_recommended_removal_file:
with open(output_recommended_removal_file, 'w') as id_f:
# 检查平均值是否大于Q3
if upstream_stats['avg'] > upstream_stats['q3'] and downstream_stats['avg'] > downstream_stats['q3']:
# 上游平均值大于其Q3,挑出上游多余碱基大于平均值的IDs
for tag, (upstream, _) in one_end_excess_counts.items():
if upstream > upstream_stats['avg']:
id_f.write(f"{tag}\n")
# 下游平均值大于其Q3,挑出下游多余碱基大于平均值的IDs
for tag, (_, downstream) in one_end_excess_counts.items():
if downstream > downstream_stats['avg']:
id_f.write(f"{tag}\n")
except Exception as e:
raise RuntimeError(f"写入多余碱基统计摘要文件时发生错误: {e}")
def write_output(sequences, file_path):
"""写入输出序列到FASTA文件"""
try:
with open(file_path, 'w') as output_file:
for tag, seq in sequences.items():
output_file.write(f">{tag}\n{seq}\n")
except Exception as e:
raise RuntimeError(f"写入输出文件时发生错误: {e}")
def process_fasta(fasta_file, output_telomere_file='telomere_output.fasta',
output_non_telomere_file='non_telomere_output.fasta',
output_one_end_file=None,
output_both_ends_file=None,
output_stat_file='reads_stats.txt',
output_excess_file=None,
output_excess_summary_file=None,
output_recommended_removal_file='recommended_removal.txt'):
sequences = read_fasta(fasta_file)
telomere_sequences, non_telomere_sequences, one_end_telomere_sequences, both_ends_telomere_sequences, one_end_excess_counts = extract_telomere_sequences(sequences)
original_sequence_count = len(sequences)
original_total_length = sum(len(seq) for seq in sequences.values())
write_output(telomere_sequences, output_telomere_file) if telomere_sequences else None
write_output(non_telomere_sequences, output_non_telomere_file) if non_telomere_sequences else None
if output_one_end_file:
write_output(one_end_telomere_sequences, output_one_end_file)
if output_both_ends_file:
write_output(both_ends_telomere_sequences, output_both_ends_file)
write_statistics(non_telomere_sequences, telomere_sequences, one_end_telomere_sequences,
both_ends_telomere_sequences, one_end_excess_counts,
output_stat_file, original_sequence_count, original_total_length)
write_excess_counts(one_end_excess_counts, output_excess_file)
write_excess_summary(one_end_excess_counts, output_excess_summary_file, output_recommended_removal_file)
def main():
"""主函数,处理命令行参数和程序入口"""
#print(script_description)
parser = argparse.ArgumentParser(description='从FASTA文件中筛选具有端粒和不具有端粒序列。',
epilog=print_colored('更详细的信息请访问: https://mengqy2022.github.io/genomics/telomere/\n','green'))
parser.add_argument('-f', '--fasta_file', type=str, help='输入FASTA文件路径', required=True)
parser.add_argument('-ot', '--output_telomere_file', type=str, help='输出端粒序列的FASTA文件路径', default='telomere_output.fasta')
parser.add_argument('-ont', '--output_non_telomere_file', type=str, help='输出非端粒序列的FASTA文件路径', default='non_telomere_output.fasta')
parser.add_argument('-oe', '--output_one_end_file', type=str, help='输出一端有端粒序列的FASTA文件路径')
parser.add_argument('-ob', '--output_both_ends_file', type=str, help='输出两端有端粒序列的FASTA文件路径')
parser.add_argument('-os', '--output_stat_file', type=str, help='输出统计信息文件路径', default='reads_stats.txt')
parser.add_argument('-odr', '--output_excess_file', type=str, help='输出多余碱基统计信息文件路径')
parser.add_argument('-ods', '--output_excess_summary_file', type=str, help='输出多余碱基统计摘要文件路径')
args = parser.parse_args()
process_fasta(args.fasta_file, args.output_telomere_file,
args.output_non_telomere_file, args.output_one_end_file,
args.output_both_ends_file, args.output_stat_file,
args.output_excess_file, args.output_excess_summary_file)
current_date = datetime.datetime.now().strftime("%Y-%m-%d %H:%M:%S")
print('Done!\n')
print('结束时间: ' + current_date + '\n')
print(f"端粒序列文件路径: {args.output_telomere_file}")
print(f"非端粒序列文件路径: {args.output_non_telomere_file}")
print(f"统计信息文件路径: {args.output_stat_file}\n")
if args.output_excess_file:
print(f"多余碱基统计文件路径: {args.output_excess_file}")
if args.output_excess_summary_file:
print(f"多余碱基统计摘要文件路径: {args.output_excess_summary_file}")
if args.output_one_end_file:
print(f"一端有端粒序列文件路径: {args.output_one_end_file}")
if args.output_both_ends_file:
print(f"两端有端粒序列文件路径: {args.output_both_ends_file}")
print('\n\t>>> mengqingyao <<<\n')
print('\t如果有任何问题请及时联系\n')
print('\t邮箱:<15877464851@163.com>\n')
if __name__ == "__main__":
main()Paste copy the above script into a file and it will work, for example:
python ./Get_tel_seq_upgrade.py
Results
Easy to use
Folder structure:
python Get_tel_seq_upgrade.py -f ./contigs.fasta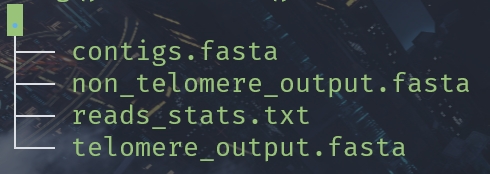
Output all files
python Get_tel_seq_upgrade.py -f ./contigs.fasta -ot telomere.fa -ont non_telomere.fa -oe one_telomere.fa -ob two_telomere.fa -os stats_reads.txt -odr stats_bases.txt -ods sum_bases.txt
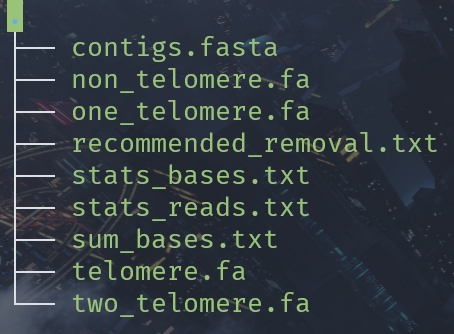
This script only used for euplotes genomics, if you want to use it for other species, please modify the code.
"telomere_start_pattern" and "telomere_end_pattern" are need to be modified for telomeres of other species.
Presentation
About the “recommended_removal.txt”, you can use it to remove the sequences with “telomeres.fasta” or not.
Email me with more questions! 584338215@qq.com